
What is Batten Disease?
Batten disease is named after the British pediatrician who first described it in 1903. Also known as Spielmeyer-Vogt-Sjogren-Batten disease, it is the most common form of a group of disorders called Neuronal Ceroid Lipofuscinoses (or NCL).
Although Batten disease is usually regarded as the Juvenile form of NCL, it has now become the term to encompass all forms of NCL.
The forms of NCL are classified by age of onset and have the same basic cause, progression and outcome but are all genetically different, meaning each is the result of a different gene. Over time, affected children suffer mental impairment, worsening seizures, and progressive loss of sight and motor skills. Eventually, children with Batten disease/NCL become blind, bedridden and unable to communicate, and, presently, it is always fatal.
Batten disease is not contagious or, at this time, preventable.
The History of Neuronal Ceroid Lipofuscinosis
The first probable instances of this condition were reported in 1826 in a Norwegian medical journal by Dr. Christian Stengel, who described three affected siblings in a small mining community in Norway. Although no pathological studies were performed on these children, the clinical descriptions are so succinct that the diagnosis of the Spielmeyer-Sjogren (juvenile) type is fully justified. More fundamental observations were reported by F.E. Batten in 1903, and by Vogt in 1905, who performed extensive clinicopathological studies on several families. Retrospectively, these papers disclose that the authors grouped together different types of the disease.
Furthermore Batten, at least for some time, insisted that the condition he described was distinctly different from Tay-Sachs Disease, the prototype of a neuronal lysosomal disorder now identified as GM2-Gangliosidosis type A. Around the same time, Spielmeyer reported detailed studies on three siblings, suffering from the Spielmeyer-Sjogren (juvenile) type, which led him to the very firm statement that this malady is not related to Tay-Sachs Disease. Subsequently, however, the pathomorphological studies of Schaffer made these authors change their minds to the extent that they reclassified their respective observations as variants of Tay-Sachs Disease, which caused confusion for about 50 years.
In 1913-14, M. Bielschowsky delineated the Late Infantile form of NCL. However, all forms were still thought to belong in the group of “familial amaurotic idiocies,” of which Tay-Sachs was the prototype.
In 1931, the Swedish psychiatrist and geneticist, Torben Sjogren, presented 115 cases with extensive clinical and genetic documentation and came to the conclusion that the disease which we now call the Spielmeyer-Sjogren (juvenile) type is genetically separate from Tay-Sachs.
Departing from the careful pathomorphological observations of Spielmeyer, Hurst, Sjovall and Ericsson, Zeman and Alpert made a determined effort to document the previously suggested pigmentary nature of the neuronal deposits in certain types of storage disorders. Simultaneously, Terry, Korey and Svennerholm demonstrated a specific ultrastructure and biochemistry for Tay-Sachs Disease, and these developments led to the distinct identification, and separation, of the NCLs from Tay-Sachs Disease by Zeman and Donahue. At that time, it was proposed that the Late Infantile (Jansky-Bielschowsky), the Juvenile (Spielmeyer-Vogt), and the adult forms (Kufs) were quite different from Tay-Sachs Disease with respect to chemical pathology and ultrastructure, and also different from other forms of sphingolipidoses. Subsequently, it was shown by Santavuori and Haltia that an Infantile form of NCL exists, which Zeman and Dyken had included with the Jansky-Bielschowsky type.
What are the forms of NCL/Batten Disease?
There are four main types of NCL, including two forms that begin earlier in childhood and a very rare form that strikes adults. The symptoms are similar but the forms become apparent at different ages and progress at different rates.
- Infantile NCL (Santavuori-Haltia disease) begins between about 6 months and 2 years of age and progresses rapidly. Affected children fail to thrive and have abnormally small heads (microcephaly). Also typical are short, sharp muscle contractions called myoclonic jerks. Initial signs of this disorder include delayed psychomotor development with progressive deterioration, other motor disorders, or seizures. The Infantile form has the most rapid progression and children live into their mid-childhood years.
- Late Infantile NCL (Jansky-Bielschowsky disease) begins between ages 2 and 4. The typical early signs are loss of muscle coordination (ataxia) and seizures along with progressive mental deterioration. This form progresses rapidly and ends in death between ages 8 and 12.
- Juvenile NCL (Batten disease) begins between the ages of 5 and 8. The typical early signs are progressive vision loss, seizures, ataxia or clumsiness. This form progresses less rapidly and ends in death in the late teens or early 20s, although some may live into their 30s.
- Adult NCL (Kufs disease or Parry disease) generally begins before the age of 40, causes milder symptoms that progress slowly, and does not cause blindness. Although age of death is variable among affected individuals, this form does shorten life expectancy.
There are six additional diseases included in the Batten disease/NCL group:
- Finnish Late Infantile – identified in Finland
- Variant Late Infantile – identified in Costa Rica, South America, Portugal and other nations
- Turkish Late Infantile – identified in Turkey
- Northern Epilepsy/ERMP – Epilepsy with Mental Retardation – identified in Finland
- Variant Juvenile – identified in Germany and USA
- Congenital/CTSD – identified in Europe
A more precise chart of the forms of Batten disease is below:
| Form | Initials | Gene | Age of Onset |
| Infantile | INCL | CLN1 | 6 mos. — 2 yrs. |
| Late Infantile | LINCL | CLN2 | 2 — 4 yrs. |
| Juvenile | JNCL | CLN3 | 5 — 7 yrs. |
| Adult | ANCL | CLN4 | 25 — 40 yrs. |
| Finnish Late Infantile | fLINCL | CLN5 | 2 — 4 yrs. |
| Variant Late Infantile | vLINCL | CLN6 | 3 — 5 yrs. |
| Turkish Late Infantile | tLINCL | CLN7 | 2 — 4 yrs. |
| Northern Epilepsy | EPMR | CLN8 | 5 — 10 yrs. |
| Variant Juvenile | vJNCL | CLN9 | 5 — 7 yrs. |
| Congenital | CTSD | CLN10 | Birth — 2 yrs. |
How many people have these disorders?
Batten disease/NCL is relatively rare, occurring in an estimated 2 to 4 of every 100,000 births in the United States, but no one really knows how many affected children there may be in North America or anywhere else in the world. The diseases have been identified worldwide. Although NCLs are classified as rare diseases, they often strike more than one person in families that carry the defective gene.
How are NCLs inherited?
Childhood NCLs are autosomal recessive disorders; that is, they occur only when a child inherits two copies of the defective gene, one from each parent. When both parents carry one defective gene, each of their children faces a one in four chance of developing NCL. At the same time, each child also faces a one in two chance of inheriting just one copy of the defective gene. Individuals who have only one defective gene are known as carriers, meaning they do not develop the disease, but they can pass the gene on to their own children.
Adult NCL may be inherited as an autosomal recessive (Kufs) or, less often, as an autosomal dominant (Parry) disorder. In autosomal dominant inheritance, all people who inherit a single copy of the disease gene develop the disease. As a result, there are no unaffected carriers of the gene.
What causes these diseases?
Symptoms of Batten disease/NCLs are linked to a buildup of substances called lipopigments in the body’s tissues. These lipopigments are made up of fats and proteins. Their name comes from the technical word lipo, which is short for “lipid” or fat, and from the term pigment, used because they take on a greenish-yellow color when viewed under an ultraviolet light microscope.
The lipopigments build up in cells of the brain and the eye, as well as in skin, muscle, and many other tissues. Inside the cells, these pigments form deposits with distinctive shapes that can be seen under an electron microscope. Some look like half-moons (or comas) and are called curvilinear bodies; others look like fingerprints and are called fingerprint inclusion bodies; and still others resemble gravel (or sand) and are called granual osmophilic deposits (GRODS).
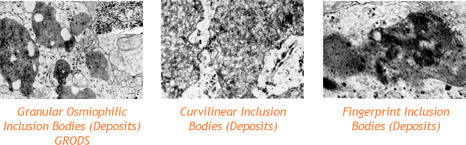
These deposits are what doctors look for when they examine a skin sample to diagnose Batten disease. The diseases cause the death of neurons (specific cells found in the brain, retina and central nervous system). The reason for neuron death is still not known.
How are these disorders diagnosed?
Because vision loss is often an early sign, Batten disease/NCL may first be suspected during an eye exam. An eye doctor can detect a loss of cells within the eye that occurs in the three childhood forms of Batten disease/NCL. However, because such cell loss occurs in other eye diseases, the disorder cannot be diagnosed by this sign alone.
Often an eye specialist/ophthalmologist or other physician who suspects Batten disease/NCL may refer the child to a neurologist, a doctor who specializes in diseases of the brain and nervous system. In order to diagnose Batten disease/NCL, the neurologist needs the patient’s medical history and information from various laboratory tests. Below are pictures of the retina showing the telltale signs of Batten disease.
Batten Disease and Ophthalmology
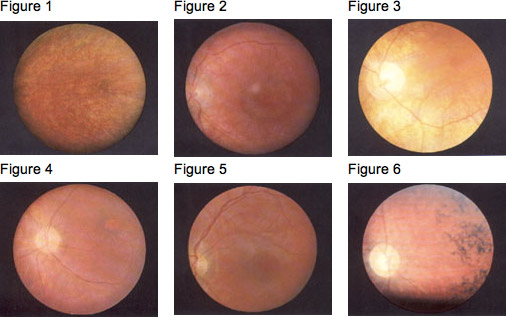
In the Fundus (the interior surface of the eye), the pigmentary changes in the macula are initially slight, and so it is easy to miss them, especially when no pupil dilation is applied and the fundus is not examined carefully. Fluorescent angiography demonstrates the pigmentary changes more clearly (Prammer, et al., 1978- ); sometimes fluorescence can be observed, leaking out of the retinal vessels. The density of the fine particuled pigmentations is slight around the macula and increases towards the periphery (Gottinger, et al., 1971- ). Dyken (1976) also mentions peripheral depigmentation. The pigment epithelium frequently has a granular “pepper and salt” appearance (see Fig. 1); sometimes there is a characteristic “bull’s eye” maculopathy (see Fig. 2, Fig. 3). The papilla becomes paler and the retinal arterioles seem more obviously constricted and extended (Fig. 4, Fig. 5). The peripheral retina varies in appearance, from normal to showing pigment-epithelial (pigmented cell layer just outside the retina) abnormalities (Spalton, et al., 1980- ). Later, peripheral pigment is often seen in the form of bone corpuscular pigment (see Fig. 6). Cataracts develop later in the course of the disease.
Diagnostic tests used for Batten disease/NCLs include:
Skin or Tissue Sampling: The doctor examines a small piece of tissue under an electron microscope. The powerful magnification of the microscope helps the doctor spot typical NCL deposits. These deposits are found in many different tissues, including skin, muscle, conjunctiva, rectal and others. Blood can also be used. See inclusion body pictures above.
Electroencephalogram or EEG: An EEG uses special patches placed on the scalp to record electrical currents inside the brain. This helps doctors see telltale patterns in the brain’s electrical activity that suggest a patient has seizures.
Electrical Studies of the Eyes: These tests, which include visual-evoked responses (VER) and electro-retinagrams (ERG), can detect various eye problems common in childhood Batten disease/NCLs.
Brain Scans: Imaging can help doctors look for changes in the brain’s appearance. The most commonly used imaging technique is computed tomography (CT), which uses x-rays and a computer to create a sophisticated picture of the brain’s tissues and structures. A CT scan may reveal brain areas that are decaying in NCL patients. A second imaging technique that is increasingly common is magnetic resonance imaging, or MRI. MRI uses a combination of magnetic fields and radio waves, instead of radiation, to create a picture of the brain.
Enzyme Assay: A recent development in the diagnosis of Batten disease/NCL is the use of enzyme assays that look for specific missing lysosomal enzymes for Infantile and Late Infantile only. This is a quick and easy diagnostic test.
Genetic/DNA Testing: Each “form” of Batten disease is the result of a different gene. Genes for eight of the ten forms have been identified. Testing for these is available for diagnosis as well as carrier and prenatal status.
Is there any treatment?
As yet, no specific treatment is known that can halt or reverse the symptoms of Batten disease/NCL. However, seizures can be reduced or controlled with anticonvulsant drugs, and other medical problems can be treated appropriately as they arise. At the same time, physical and occupational therapy may help patients retain function as long as possible.
BDSRA helps scientists by fostering awareness, promoting more research, providing samples and information, and by funding research that is directed towards understanding all forms of Batten disease and development of therapies.
Support and encouragement can help children and families cope with the profound disability and losses caused by NCLs. The Batten Disease Support and Research Association enables affected children, adults and families to share common concerns and experiences.
Meanwhile, scientists pursue medical research that will someday yield an effective treatment.